Title:Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome.
doi: 10.1038/nature16192
Abstract: Roots and leaves of healthy plants host taxonomically structured bacterial assemblies, and members of these communities contribute to plant growth and health. We established Arabidopsis leaf- and root-derived microbiota culture collections representing the majority of bacterial species that are reproducibly detectable by culture-independent community sequencing. We found an extensive taxonomic overlap between the leaf and root microbiota. Genome drafts of 400 isolates revealed a large overlap of genome-encoded functional capabilities between leaf- and root-derived bacteria with few significant differences at the level of individual functional categories. Using defined bacterial communities and a gnotobiotic Arabidopsis plant system we show that the isolates form assemblies resembling natural microbiota on their cognate host organs, but are also capable of ectopic leaf or root colonization. While this raises the possibility of reciprocal relocation between root and leaf microbiota members, genome information and recolonization experiments also provide evidence for microbiota specialization to their respective niche.
At-RSPHERE and At-LSPHERE culture collections
Phylogenetic analysis based on 16S rRNA gene Sanger sequences revealed that 119 out of 206 At-RSPHERE isolates (58%) share ≥97% sequence identity matches with corresponding 16S rRNA gene fragments of At-LSPHERE members (outermost circle in Fig. 1a). Similarly, 108 out of 224 At-LSPHERE isolates (48%) share ≥97% sequence identity matches with At-RSPHERE members (outermost circle in Fig. 1b).
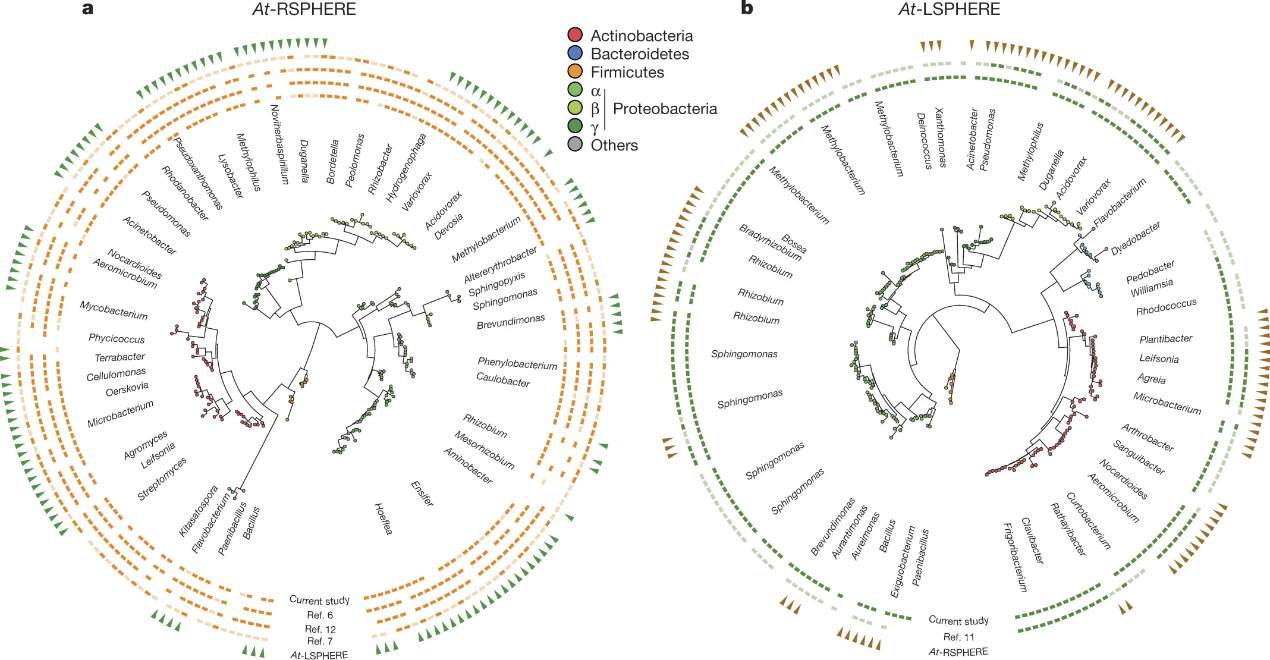
Figure 1: Taxonomic overlap between At-RSPHERE and At-LSPHERE isolates and their representation in culture-independent microbiota profiling studies.
a, b, Phylogenetic trees of At-RSPHERE (a; n = 206 isolates) and At-LSPHERE (b; n = 224 isolates) bacteria. Their taxonomic overlap is shown in the outermost ring (green or brown triangles). a, Representation of At-RSPHERE bacteria in each of four indicated culture-independent profiling studies of the A. thaliana root microbiota; root-associated OTUs with RAs ≥0.1% (dark orange) or ≤0.1% (light orange). b, Representation of At-LSPHERE bacteria in the two indicated culture-independent phyllosphere profiling studies; leaf-associated OTUs with RAs ≥0.1% (dark green) or <0.1% (light green). Taxonomic assignment and phylogenetic tree inference were based on partial 16S rRNA gene Sanger sequences.
Comparative genome analysis of the culture collections
To characterize the functional capabilities of the core culture collections we subjected each isolate to whole-genome sequencing and generated a total of 432 high-quality draft genomes (206 from leaf, 194 from root and 32 from soil; ). Taxonomic assignment of the whole-genome sequences confirmed that these isolates span a broad taxonomic range, belonging to 35 different bacterial families distributed across five phyla.
We then examined the functional diversity between the sequenced isolates in order to determine whether the observed phylogenetic overlap corresponded with functional similarities between leaf and root isolates. Principal coordinates analysis (PCoA) of functional distances (Fig. 2a;) revealed a clear clustering of genomes on the basis of their taxonomy, but only limited separation of genomes on the basis of their ecological compartment. Taken together, both phylogenetic and functional diversification of the genomes is strongly driven by their taxonomic affiliation and weakly by the ecological niche.
We examined the functional diversity within each bacterial family (Fig. 2b) in order to identify bacterial taxa with varying degrees of functional versatility. Families belonging to Actinobacteria show a lower functional diversity (average distance 0.37) compared to those belonging to Bacteroidetes, Firmicutes and especially Proteobacteria (0.65 average pair-wise distance), which exhibit a higher degree of within-family functional diversification, even though all family-level groups have a comparable degree of phylogenetic relatedness. Among these groups, Pseudomonadaceae, Oxalobacteraceae and Methylobacteriaceae members show the highest functional heterogeneity, compared to Microbacteriaceae strains, which we identified as the least functionally diverse family (Fig. 2b).
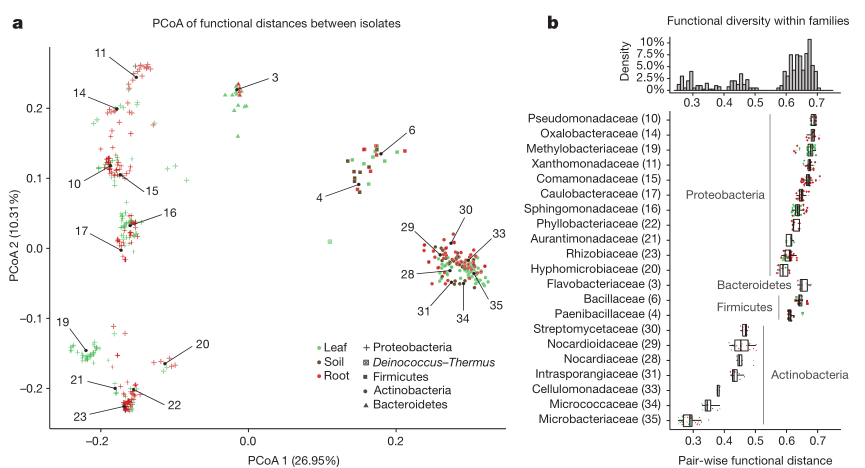
Figure 2: Analysis of functional diversity between sequenced isolates.
a, Principal coordinate analysis (PCoA) plot depicting functional distances between sequenced genomes (n = 432) based on the KEGG Orthology (KO) database annotation. Each point represents a genome. Colours represent the organ of isolation and shapes correspond to their taxonomy. Numbers inside the plot refer to bacterial families listed in b. b, Analysis of functional diversity within bacterial families as measured by pair-wise functional distances between genomes (bottom panel; n = 432). Higher pairwise distances between members of a family indicate a larger degree of functional diversity. Only families with at least five members are shown. The histogram (top panel) was calculated for the entire data set and the y-axis corresponds to the percentage of data points in each bin. Boxplot whiskers extend to the most extreme data point which is no more than 1.5 times the interquartile range from the upper or lower quartiles.
Specifically, we found the category ‘carbohydrate metabolism’ to be enriched in the leaf and soil genomes compared to those isolated from roots (Mann–Whitney test, P = 1.29 × 10−7; Fig. 3b). We speculate that this differential enrichment could reflect the availability of simple carbon sources in roots through the process of root exudation (sugars, amino acids, aliphatic acids), whereas bacteria associated with leaves or unplanted soil might rely on a more diverse repertoire of carbohydrate metabolism genes to access scarce and complex organic carbon, for example, polysaccharides and leaf cuticular waxes. The category ‘xenobiotics biodegradation and catabolism’ is enriched in the root genomes with respect to those isolated from leaves (P = 2.60 × 10−11; Fig. 3b), which is consistent with previous evidence that genes for aromatic compound utilization are expressed in the rhizosphere.
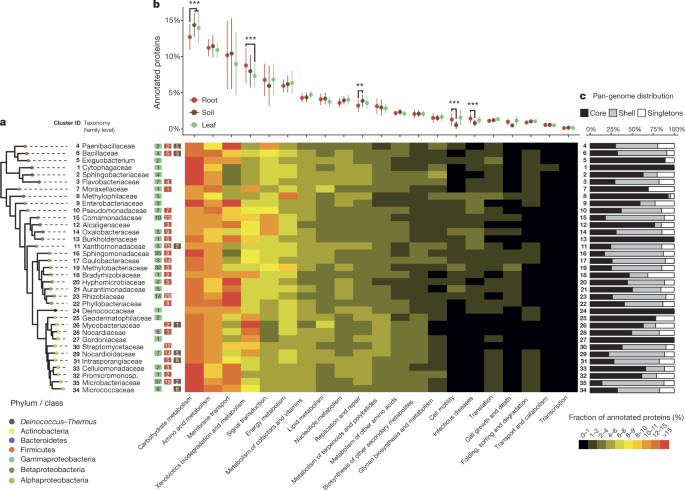
Figure 3: Functional analysis of sequenced isolates.
a, Phylogeny of family-level clusters of bacterial isolates. The tips of the tree are annotated, from left to right, with the cluster ID, taxonomic classification, followed by the number of sequenced isolates from leaf, root or soil that constitute each cluster. The heat map depicts the average percentage of annotated proteins of each cluster belonging to each functional category. b, Functional enrichment analysis between leaf (n = 206), root (n = 194) and soil (n = 32) genomes. Points and bars correspond to the mean abundance and standard deviation of each functional category. P values were obtained using the non-parametric Mann–Whitney test corrected by the Bonferroni approach. c, Analysis of pan-genome distribution for each cluster of genomes, indicating the percentage of annotated proteins found in only one isolate (singletons), in more than one but not all (shell) or in all genomes within the cluster (core).
Synthetic community colonization of germ-free plants
To mimic the taxonomic diversity of leaf and root microbiota in natural environments we employed mainly two SynComs: ‘L’ comprising 218 leaf-derived bacteria and ‘R+S’ consisting of 188 members of which 158 are root-derived and 30 are soil-derived bacteria.
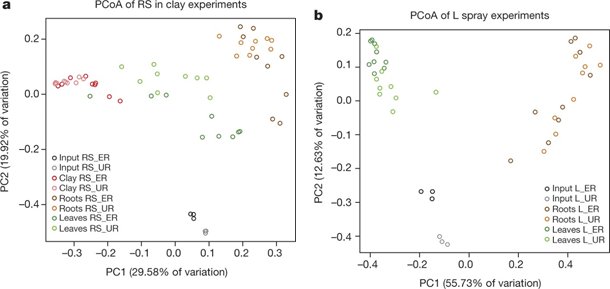
Figure 4: SynCom colonization of germ-free A. thaliana plants.
a, b, Principal coordinate analysis (PCoA) of Bray–Curtis distances of input and output SynCom profiles of RS in clay (a; n = 60) and L spray (b; n = 42) experiments. Each condition was tested with 6 independently prepared SynComs; each preparation was used for 3 independent inoculations. L, leaf-derived strains; RS, root- and soil-derived strains; ER, equal strain ratio; UR, unequal strain ratio.
Niche-specific microbiota establishment with SynComs
The species overlap between root and leaf microbiota and their corresponding culture collections prompted us to test whether R+S and L SynComs equally contribute to root and leaf microbiota establishment. Both SynComs were pooled and inoculated in clay together with surface-sterilized A. thaliana seeds (designated ‘RSL in clay’, Fig. 5a). We also tested whether a preformed root-associated community can interfere with leaf-associated community establishment. After three weeks of co-cultivation, half of the plants grown with the ‘RSL in clay’ SynCom were treated by leaf-spray inoculation with the L SynCom supplemented with 15 root-derived strains (designated ‘RSL in clay & L+15R spray’). Plant organ-specific output communities were determined after a further four weeks of co-incubation. We also inoculated the L SynCom alone in clay and determined output SynComs (designated ‘L in clay’, Fig. 5a).
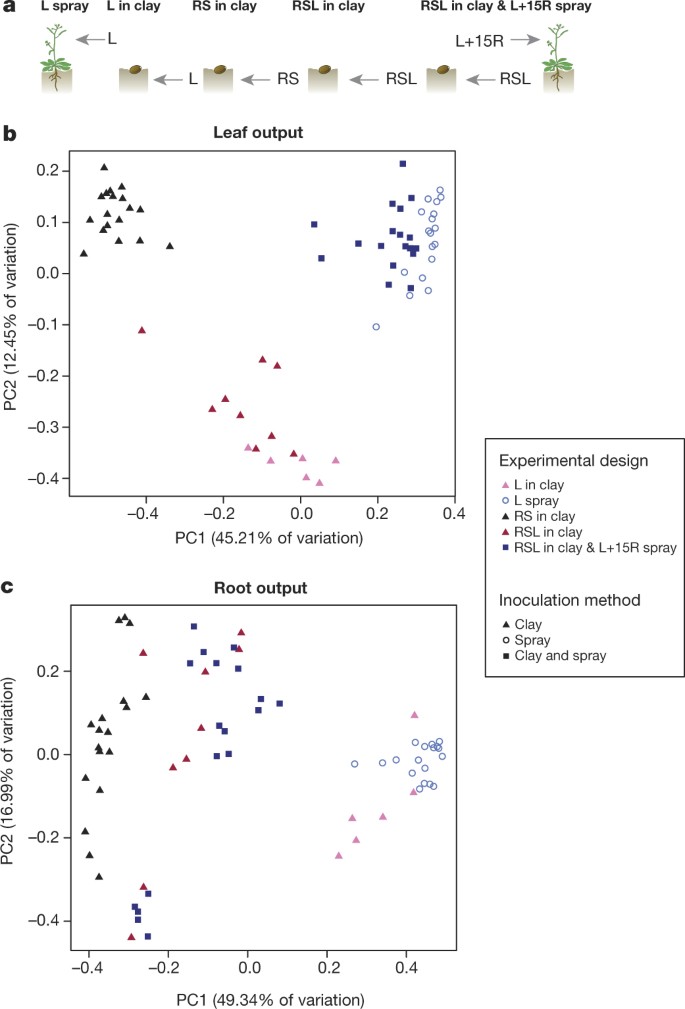
Figure 5: SynCom competition supports host-organ-specific community assemblies.
a, Pictograms illustrating ‘L spray’, ‘L in clay’, ‘RS in clay’, ‘RSL in clay’, and ‘RSL in clay & L+15R spray’ SynCom experiments. b, c, PCoA of Bray–Curtis distances of leaf (b; n = 69) and root (c; n = 69) outputs of the five experiments illustrated in a. R, root-derived isolates; S, soil-derived isolates; L, leaf-derived isolates. L in clay was tested with 6 independently prepared SynComs; RSL in clay experiment was tested with 3 independently prepared SynComs, each used for 3 independent inoculations. All other experiments were tested with 6 independently prepared SynComs and each preparation was used for 3 independent inoculations.
Conclusions
By employing systematic bacterial isolation approaches, we established expandable culture collections of the A. thaliana leaf- and root-associated microbiota, which capture the majority of the species found reproducibly in their respective natural communities (≥0.1% relative abundance). These resources together with the remarkable reproducibility of the gnotobiotic reconstitution system enable future studies on bacterial community establishment and functions under laboratory conditions.
From: He Weihua